JustOrthologs: A Fast, Accurate, and User-Friendly Ortholog-Identification Algorithm
Abstract:
Orthologous gene identification is fundamental to all aspects of biology. Specifically, ortholog identification between organisms can provide functional insights for genes of unknown function. Currently, most ortholog identification algorithms require all-versus-all Basic Local Alignment Search Tool (BLAST) comparisons, which are time consuming and memory intensive. We developed a novel algorithm, JustOrthologs, which exploits a previously unidentified bias in coding sequence (CDS) length. By requiring the same CDS lengths between two genes, JustOrthologs decreases ortholog identification runtime by 90%, while obtaining comparable precision and recall scores to OrthoMCL, OMA, and OrthoFinder. Using this technique, we compared 304 complete eukaryotic genomes and confirmed gene annotations for 148,352 genes, clustered 85,660 genes in previously unreported groups, and identified 1,737 potentially mislabeled genes across 7,110 ortholog groups.
What is an ortholog?
Orthologous genes are present in two or more species because they were derived from the same ancestral gene.
Why are orthologs important?
Orthologs provide information about the evolutionary history of species. Because orthologous genes come from the exact same gene, divergences in nucleic sequences can show selective pressure and help identify gene function. Furthermore, orthologs are typically used for phylogenetic tree reconstruction, for which genetic differences help predict time of divergence, as well as species relatedness.
Are other ortholog identification algorithms available?
Yes. However, other algorithms suffer from poor documentation, difficult usability, slow runtime, lack of precision and/or poor recall.
How does JustOrthologs compare to other algorithms?
JustOrthologs is by far the best ortholog identification algorithm. Not only is it well documented and designed to be an easy 1-step process, but its runtime is much faster than all other algorithms while achieving a higher combined precision and recall score. Take a look at the results:
Human versus bonobo runtime for datasets with some orthologs present:
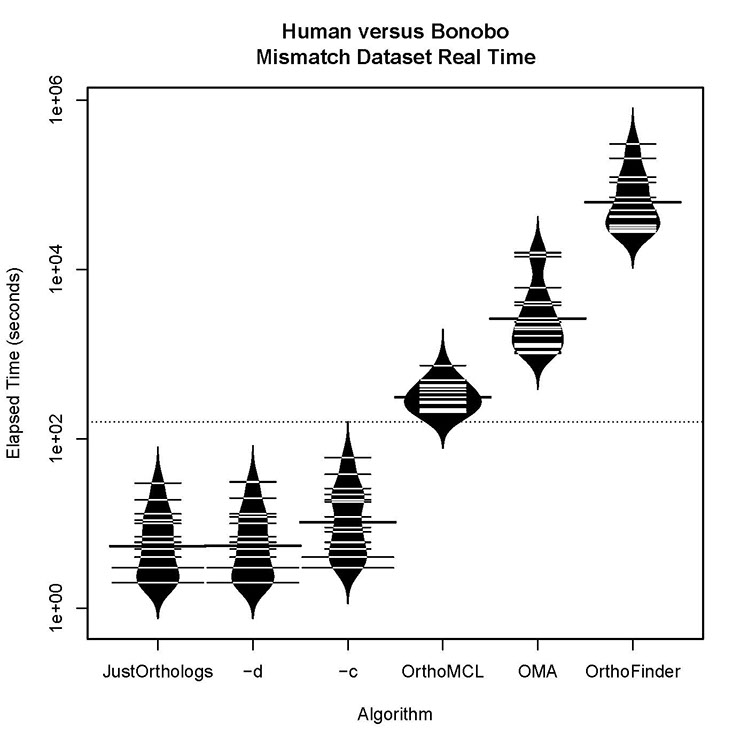
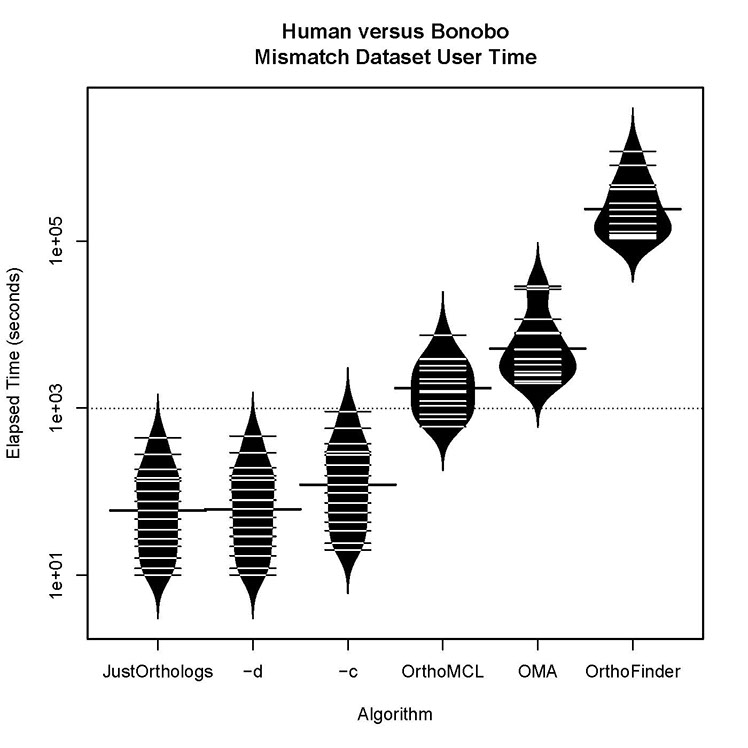
All three versions of JustOrthologs complete all computations for the datasets at least an order of magnitude faster than the other algorithms. When considering the real time, the difference is even greater because the multiprocessing capabilities of JustOrthologs outperforms those of each of the other algorithms.
Human versus bonobo accuracy for datasets with some orthologs present:
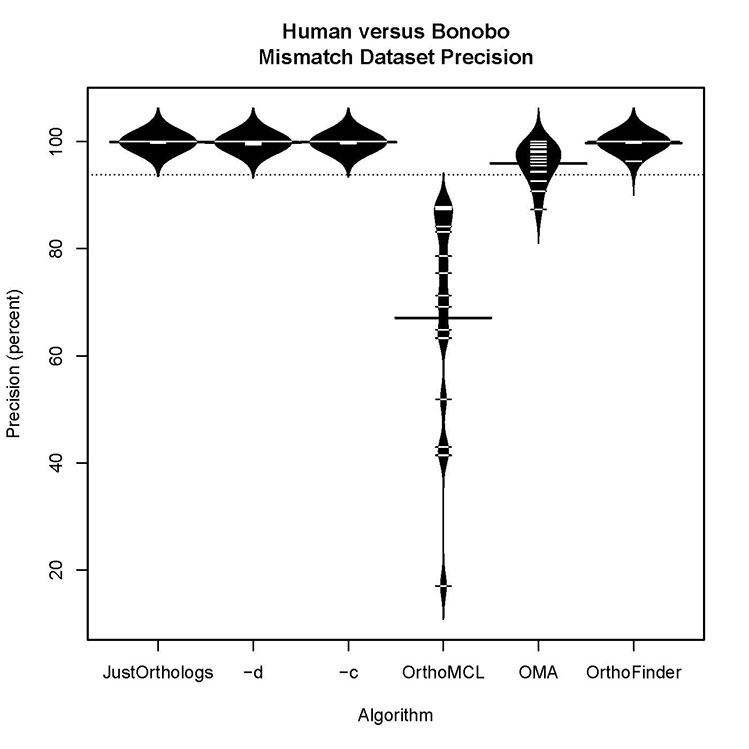
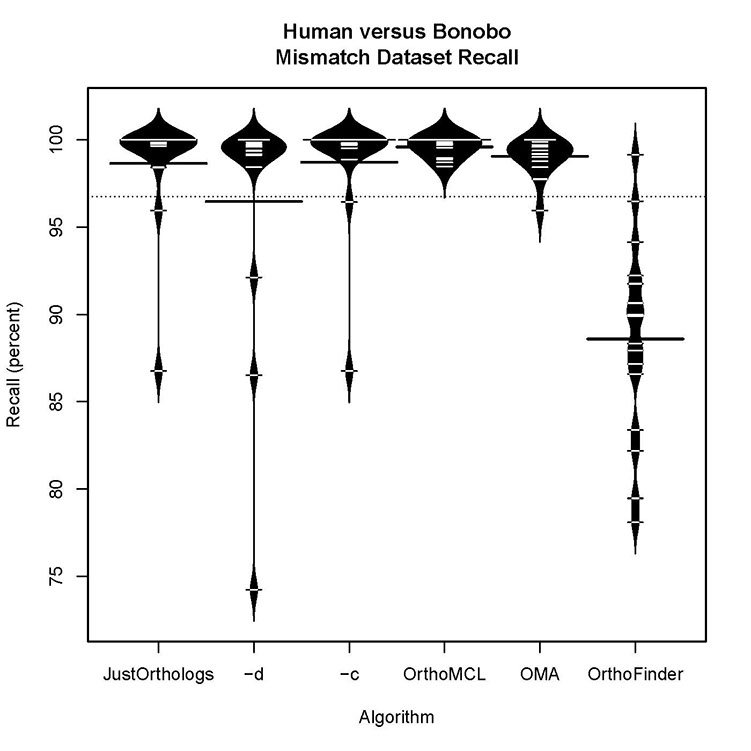
All three versions of JustOrthologs have comparable precision and accuracy scores for datasets that contained some orthologs and some false positives.
What is the false positive rate for the algorithm with no orthologs present?
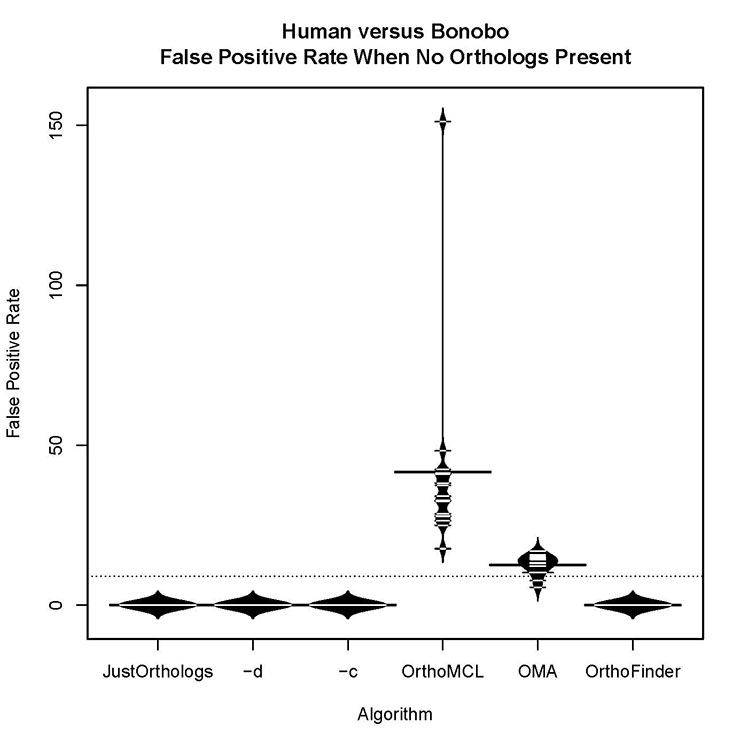
In this dataset, the false positive rate was zero. Across all datasets, the false positive rate for JustOrthologs was nearly zero, and performed significantly better than OMA or OrthoMCL.
Okay, I'm convinced that JustOrthologs is better; but how does it work?
JustOrthologs exploits a coding sequence length bias that exists between orthologs. Many genes are formed by small coding sequences being "spliced" together to form a larger gene. We discovered that the length of these coding sequences is conserved over time, meaning that we only needed to compare the sequence identity of coding sequences that were exactly the same length. By limiting the number of comparisons made, we were able to significantly reduce the runtime of the algorithm while maintain the fidelity of our results.
Conclusion:
If you have a fully annotated genome, it is advantageous to use the coding sequence lengths in JustOrthologs because it decreases runtime and increases overall accuracy. As the number of complete genome annotations increases, we believe that JustOrthologs will play a larger role in ortholog identification because of its speed and accuracy.