CODON AVERSION MOTIFS:
An alignment-free method to recover phylogenies
Abstract:
Codon bias refers to the non-random usage of synonymous codons, and differs between organisms, between genes, and even within a gene. We previously identified a strong phylogenetic signal, based on codon usage preferences, in 72 tetrapod species, focusing on stop codon usage preferences. Here we report the expansion of our previous work into >20,000 species across all kingdoms of life, and the development of tools to streamline phylogenetic inference based on codon usage preferences, and here specifically codon non-usage (or codon aversion). For each organism, we constructed a set of tuples, where each tuple contains a list of unused codons for a given gene. We define the pairwise distance between two species, A and B, as the ratio of direct overlaps to total possible overlaps. Total possible overlaps is the number of tuples in the set, for A or B, containing the fewest tuples, and direct overlaps is the intersection of tuples in the two sets. This approach allows us to calculate pairwise distances, even though there are substantial differences in the number of genes for each species. Finally, we use neighbor-joining to recover phylogenies. Using the Open Tree of Life and NCBI Taxonomy Database as expected phylogenies, our approach compares well, recovering phylogenies that largely match expected trees. Key Words: [Codon usage bias, phylogeny, codon aversion motif, species relationship, phylogenetically informative character]
Methods:
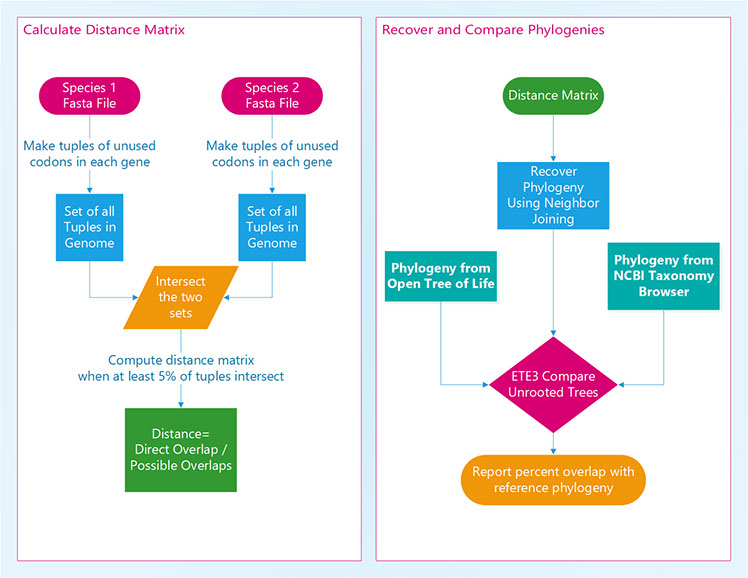
We are currently in the process of writing Codon Aversion Motifs, and it should be completed shortly please return to this page for an update on the progress of this manuscript. Feel free to explore the rest of this website and view the other works which we have published.